Radical Halogenation of Alkanes
In this tutorial, we are going to talk about the radical halogenation of alkanes. We’ll go over the intricacies of the mechanism, how to find the major products in this reaction, and discuss the most important points of each mechanistic step.
Introduction to Radical halogenation
Here are a couple of simple examples of this reaction. In each case, we replace one of the hydrogens in our alkane with the corresponding halogen. Also, we’re producing the hydrogen halide as a side product in this reaction.
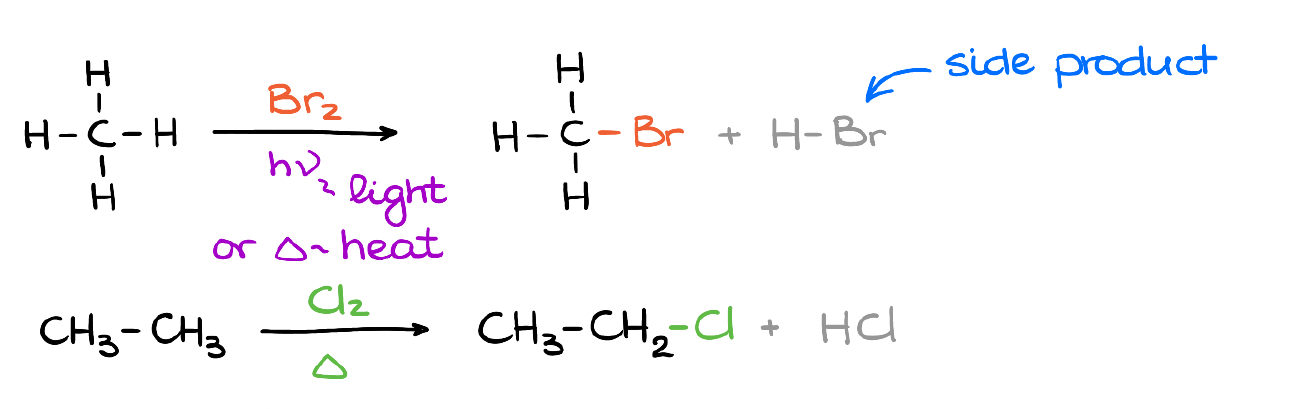
And here’s a slightly more complicated example, where we have multiple potential places for the halogen to substitute the H. Notice, that although it may look like you’re just “adding” a halogen to your molecule when we use the skeletal structures, in reality, we’re replacing one of the hydrogens that was in that position.
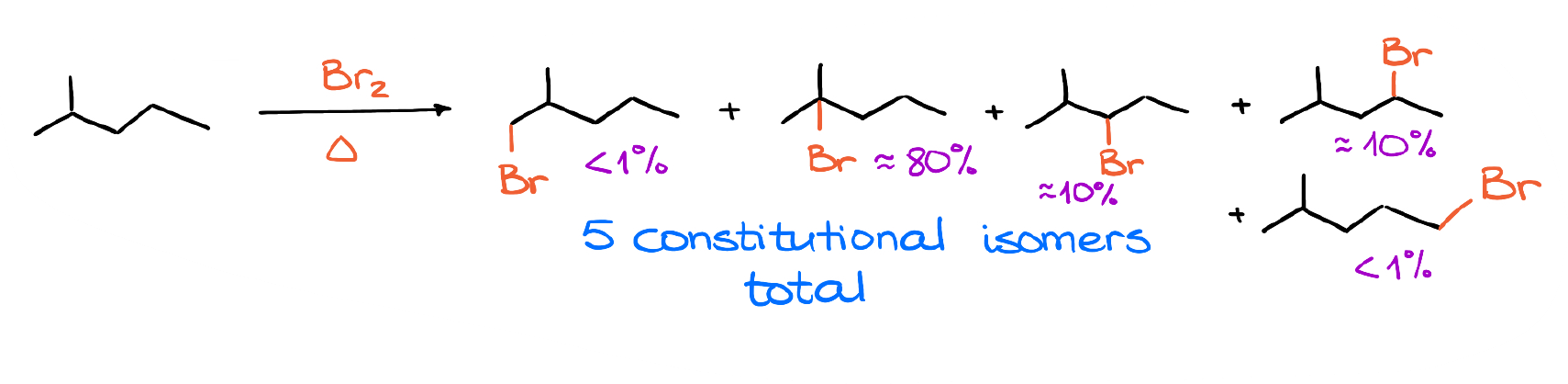
Also, while we can potentially get all the products I have here for this example, in reality we are going to have one major product, a couple of minor ones, and some traces of for the rest. But before we discuss why that’s the case, let’s start by looking at the mechanism of the radical halogenation.
Mechanism of the Radical Halogenation
Unlike many other mechanisms in organic chemistry, this one is going to have a few unique features.
The first step is the initiation step. We make new radicals in this step where we had no radicals before.
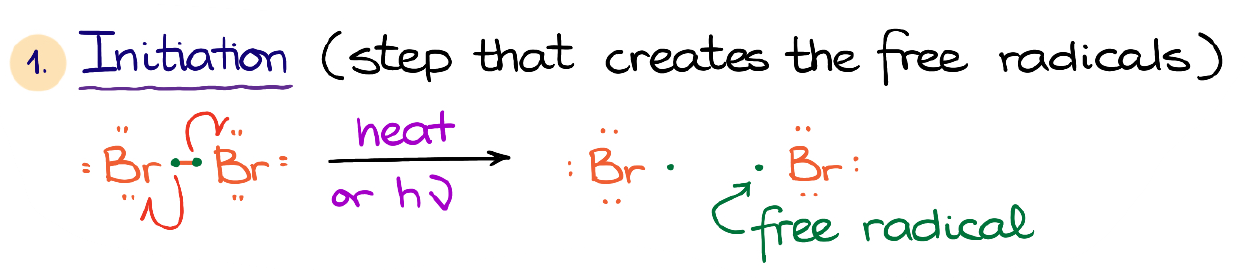
Typically, we’ll have the halogen that we want to use in this reaction split into two radicals upon heating it or using the high intensity light. This forms a couple of free radicals.
A free radical is a neutral species that has an unpaired electron on its valence shell. And since the species with an incomplete valence shell are not stable, free radicals are going to be quite reactive.
Now, why exactly does the halogen “split” into two radicals?
The Halogen-Halogen bond is relatively weak compared to the other bonds that we see in typical organic molecules. So, for instance, the Br-Br bond is almost twice weaker than the C-C bond.
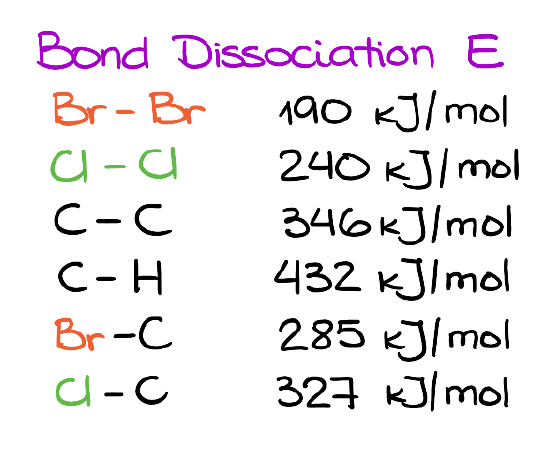
This is a general trend you’re going to see with many other bonds where you have two electronegative elements like oxygen or halogens bond to each other with a single bond. Most of those bonds are quite weak and will tend to break easily upon heating or irradiation with light.
Once we have our free radicals in the system, we’re going to start the propagation cycle which consists of two steps.
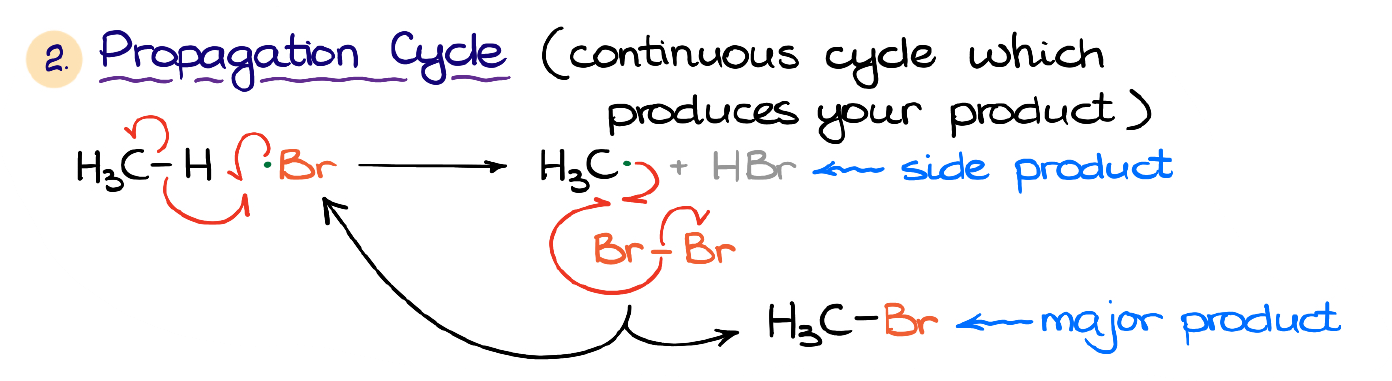
The first step is the halogen radical abstracting the hydrogen from our alkane. We form our side product (HBr in this case) in this step.
Next, we are going to have a reaction with another molecule of the halogen. This step generates the halogenated product and regenerates the halogen radical.
You may wonder why we don’t just react our organic radical with another Br radical, right? The reason for this is because we make a very low concentration of those Br radicals in the initiation step. So, it’s highly unlikely that our organic radical meets one of those Br radicals in the solution. It’s not entirely impossible, but the probability of it is extremely low. Thus, the propagation works as an engine constantly cycling through the steps of the hydrogen abstraction by one Br radical and regeneration of the new Br radical.

Eventually, we’ll get to the point, where some of those radicals will meet each other in space. At that point, we’ll have a termination step. So, the termination step is literally two random radicals hitting each other and recombining in space. This step can give you a lot of interesting molecules. However, as I’ve mentioned a moment ago, we have a very low concentration of those radicals in our solution at any given time, so the chances of them meeting are low.
This also means that although you can get the product of your reaction through the termination step, you should NEVER show the formation of the final product via the termination step on the test! The majority of your product always comes from the propagation cycle.
Energetics of the Radical Halogenation Steps
Alright, now when we know all the steps in the radical halogenation mechanism, let’s look at the energetics of each of those steps.
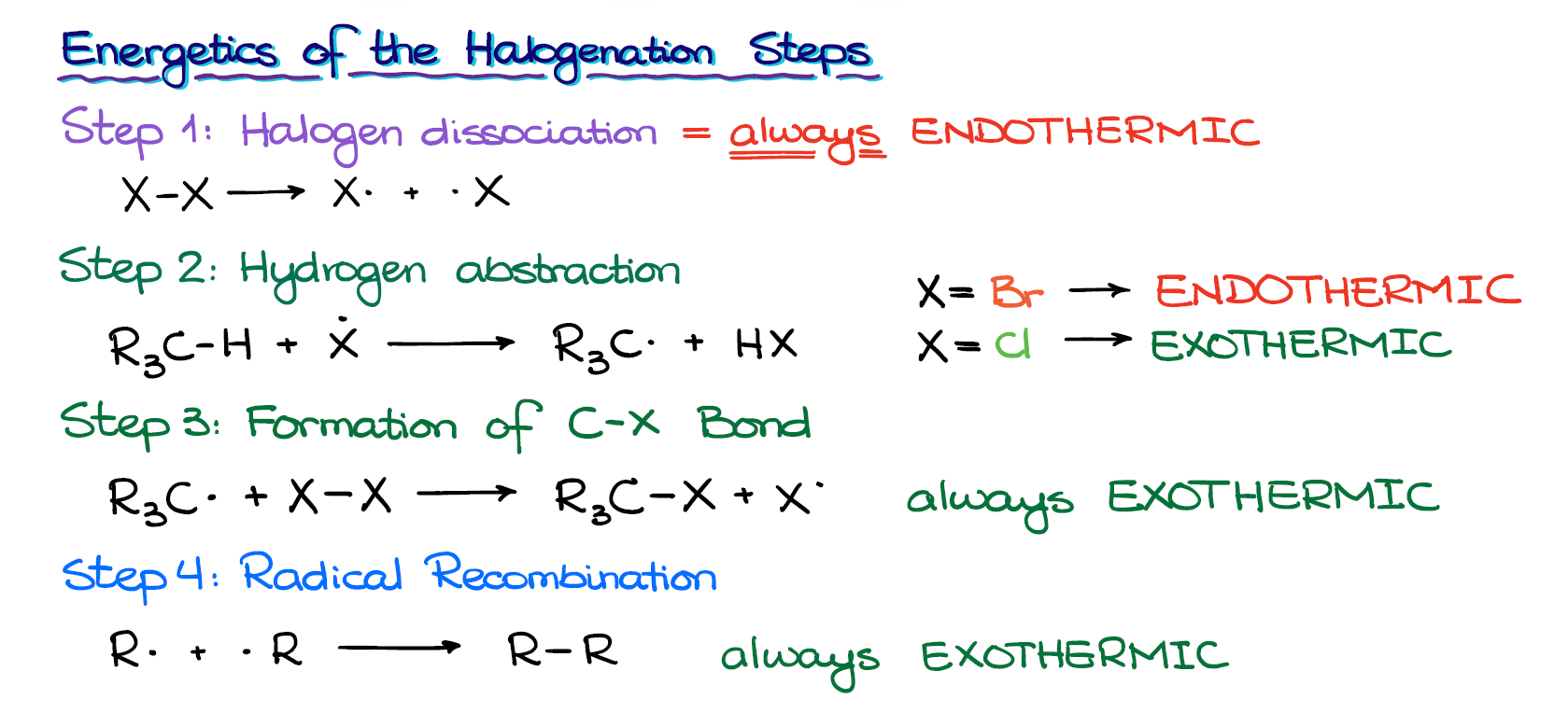
The first step, the halogen dissociation is always an endothermic process. Any bond breaking process is always endothermic! I repeat, bond breaking is ALWAYS endothermic.
The next step, however, the hydrogen abstraction—well, that one can be either endo- or exothermic depending on the halogen we have in our reaction.
When we have a bromination the reaction is endothermic. While the chlorination is the exothermic process.
The last two steps, the formation of a new C-Halogen bond and the termination steps are going to be exothermic in either case.
So, why is this important?
Regioselectivity of the Radical Halogenation Reaction
The reason why we look at the reaction energetics is to understand the overall reaction energy profile and how it may influence the reaction.
So, in the case of bromination, the formation of the radical is the rate-determining step, the slowest step in our reaction. And it has the highest activation energy. While in the case of chlorination, the formation of the radical is not the most energy-intensive step.
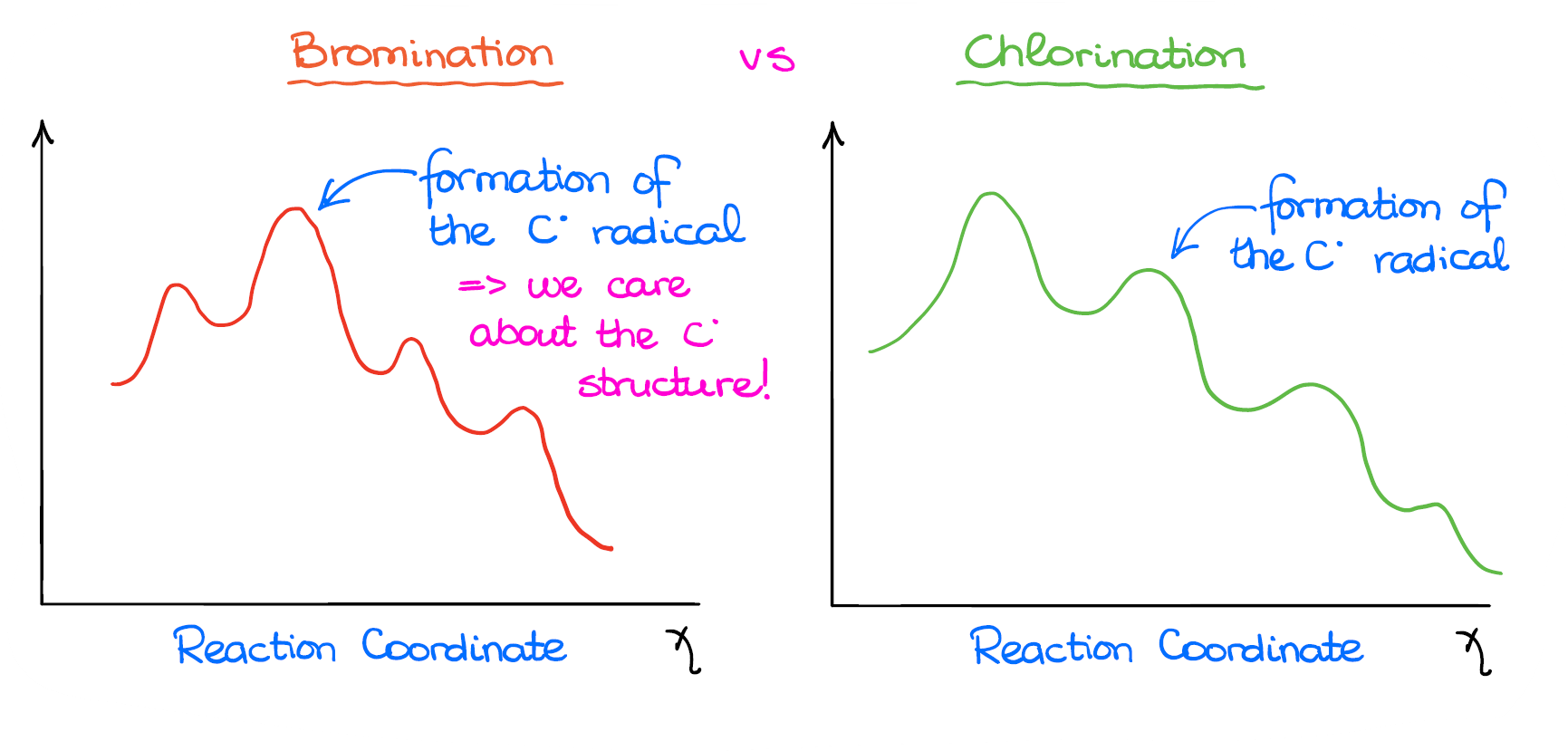
This means that in bromination, the structure and the stability of the radical are quite important. While in the chlorination, it has a much less of an impact on the reaction outcome.
This plays a huge role in the regioselectivity of the radical halogenation.
Let’s look at the example like what I showed to you at the very beginning of this tutorial.
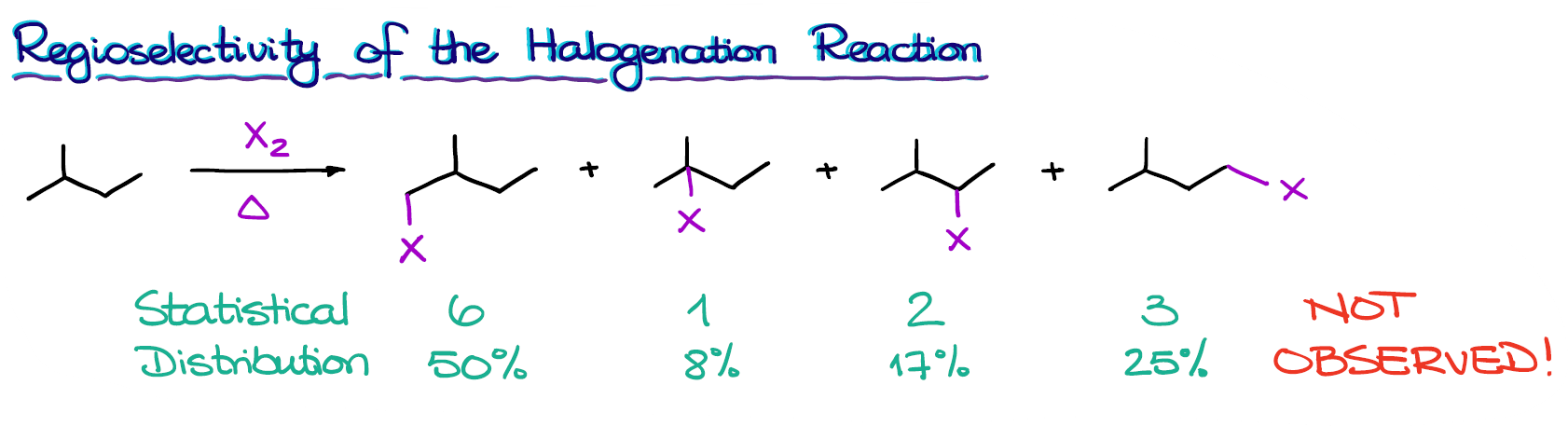
Here we have a molecule, that upon halogenation, can give you several different constitutional isomers. If the reaction proceeds in a purely statistical manner, we are going to see the following distributions of the products:
Where each number corresponds to the number of hydrogens in each position that can be substituted with a halogen. We have 6 in the first case, because we have two identical methyl groups. The rest of our hydrogen-containing groups are all unique though.
We can also represent this distribution in percentages.
The reality is a little bit more interesting. In reality, this is not what we actually observe when we perform this reaction!
So, what’s the deal here?
Well, first, it’s the radical stability. Since we have a radical intermediate that we form in the propagation step, we need to look at its stability. The resonance-stabilized radicals, like benzylic or allylic are the most stable. Then, we have the tertiary radical, which is more stable than secondary, which is more stable than primary.
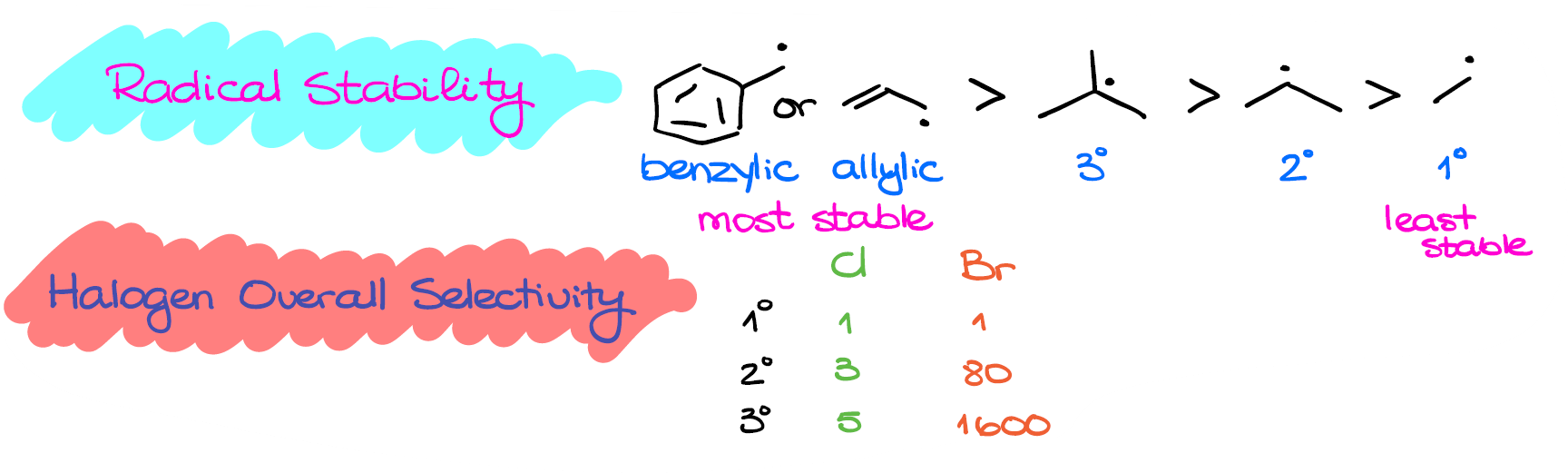
The intermediate stability is something that we generally want to consider in a reaction because the more stable the intermediate, the lower the activation energy in the reaction. And since the activation energy is inversely proportional to the reaction rate, the lower the activation energy, the faster the reaction. Thus, formation of tertiary radicals proceeds faster than the formation of secondary or primary ones.
This, together with the energetics of the steps that we discussed a moment ago explains the average halogen selectivity towards different positions in the molecule. Or, in other words, the regioselectivity of the reaction.
So, chlorine is about 3 times more selective towards the secondary carbons and about 5 times more selective towards the tertiary ones. Or you can think about it as in the chlorination reaction, the secondary radicals form 3 times faster, and the tertiary radicals form 5 times faster than the primary radicals.
When it comes to the bromination though, we have a drastically different picture. Bromine is approximately 80 times more selective towards the secondary position and about 1600 times more selective towards the tertiary position. Now, it doesn’t mean that bromination is a faster reaction, it’s overall slower. These numbers are the relative rates for each type of reaction, so we cannot compare them between.
So, how does this play into the regioselectivity?
If we now take the statistical probabilities that we already know by looking at the number of hydrogens in each position and multiply those by the relative reaction rates for each halogen, we’ll get a more realistic distribution.
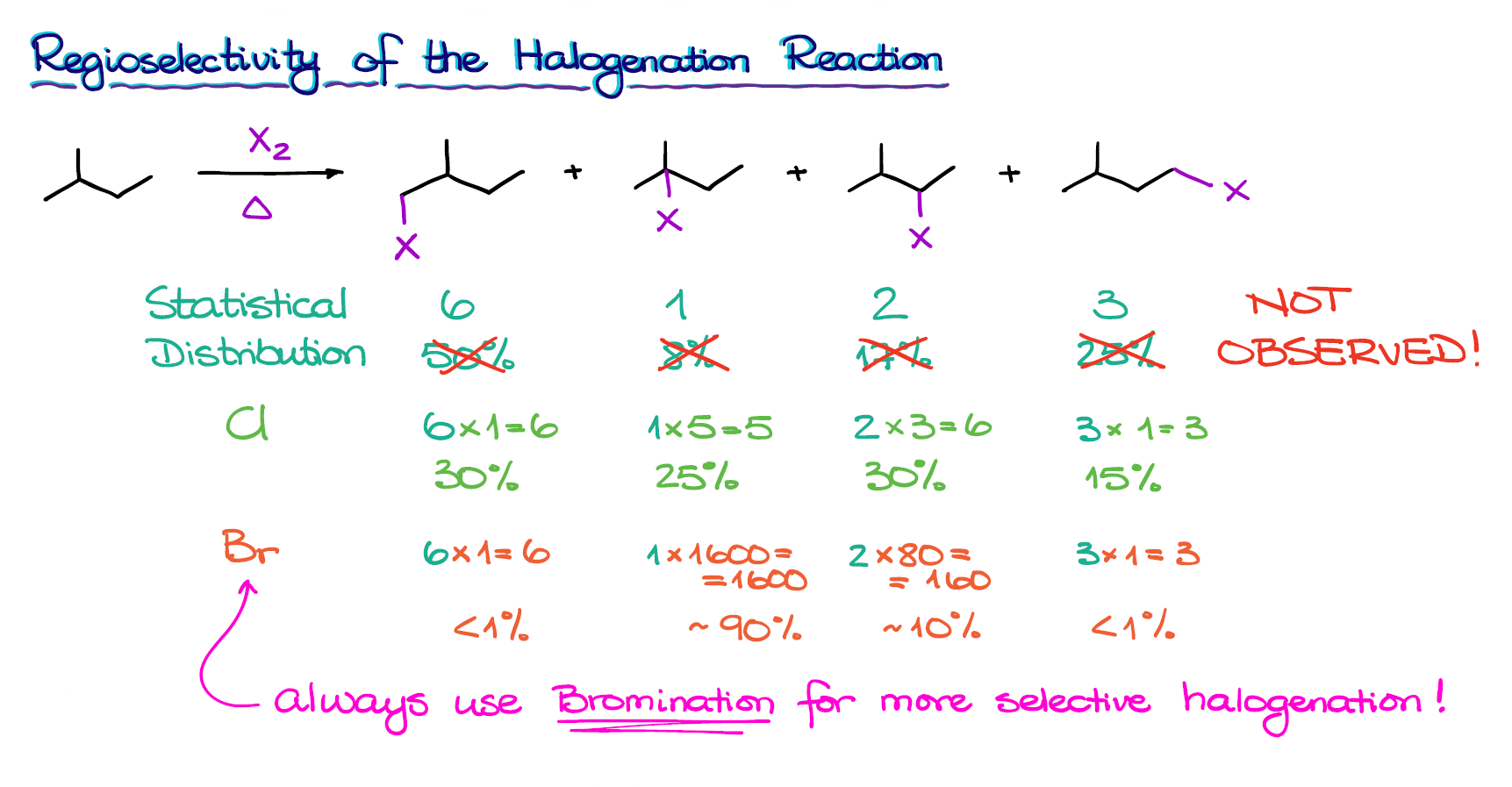
Thus, for a reaction with chlorine, we’ll get approximately 25% of the tertiary product instead of 8% compared to the purely “statistical” substitution scenario.
If we do the same manipulation for the bromination, however, we get hugely different numbers. For the reaction with bromine, we get almost 90% of the tertiary product and just a little bit of the rest.
Thus, we can say that bromine is a very regioselective reagent, while chlorine is not.
So, when you’re doing this reaction on the test, remember, that in the bromination reaction you’re always going to go after the tertiary halide as your major product. While, in the chlorination reaction, all possible products will form to an appreciable extent and the tertiary chloride may not even be the major product.
Example of the Radical Halogenation
Now, let’s look at a few more examples here.
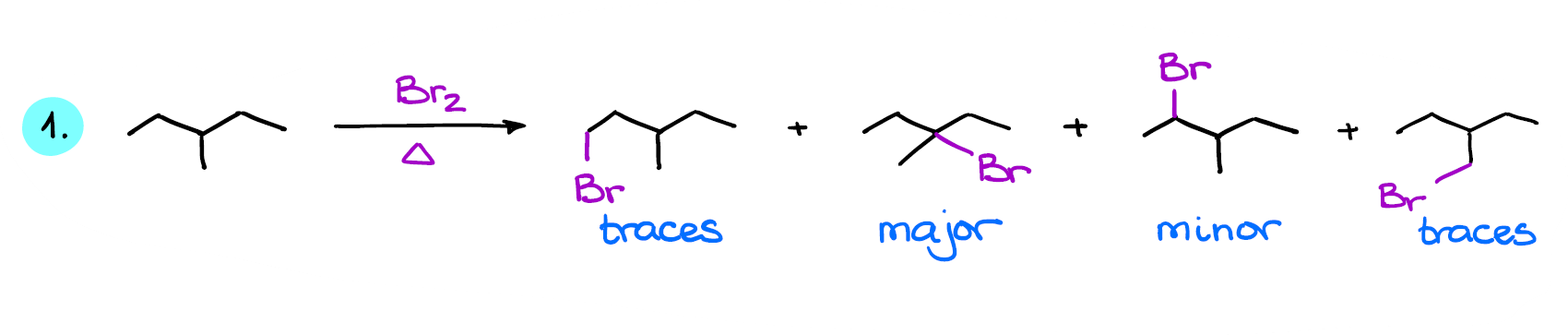
Looking at my first reaction here, I can see that I have three different positions where the substitution can occur. Due to the internal symmetry in this molecule, the right and the left side of the molecule are the same.
This way I can quickly draw my three possible products.
Also, since this is a bromination reaction, we can easily say that the tertiary bromide is going to be the major product in this reaction.
Here’s another example. This molecule also has some symmetry, so some of the positions around the molecule will be identical.
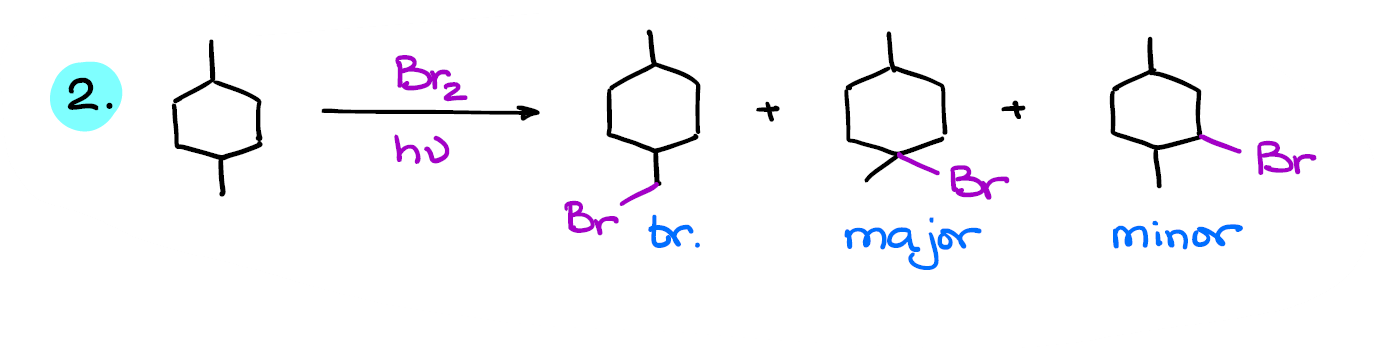
So, we have 4 possible products in this case. And again, the tertiary product is major in this reaction.
And finally, let’s look at this example.
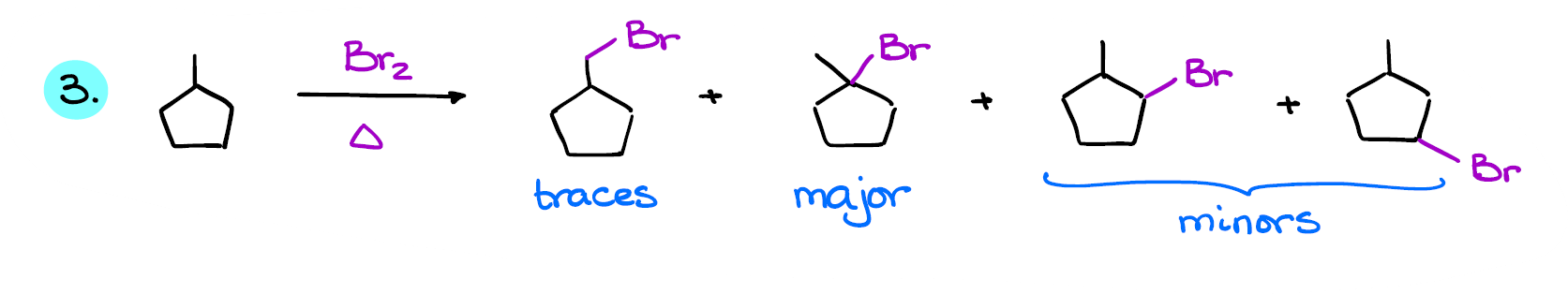
Here we have 4 possible products. And like in the previous case, since it’s a bromination, the retiary halide is going to be the major product in this case as well.
I purposefully chose the bromination for these three examples as it’s a selective process and it’s the most common type of halogenation you’re going to see on the test. In most cases, even if you see chlorination, your instructor will still expect you to show the tertiary product as the major product unless they went through the details of the regioselectivity for each halogen like we did here.